Optimization of docking parameters
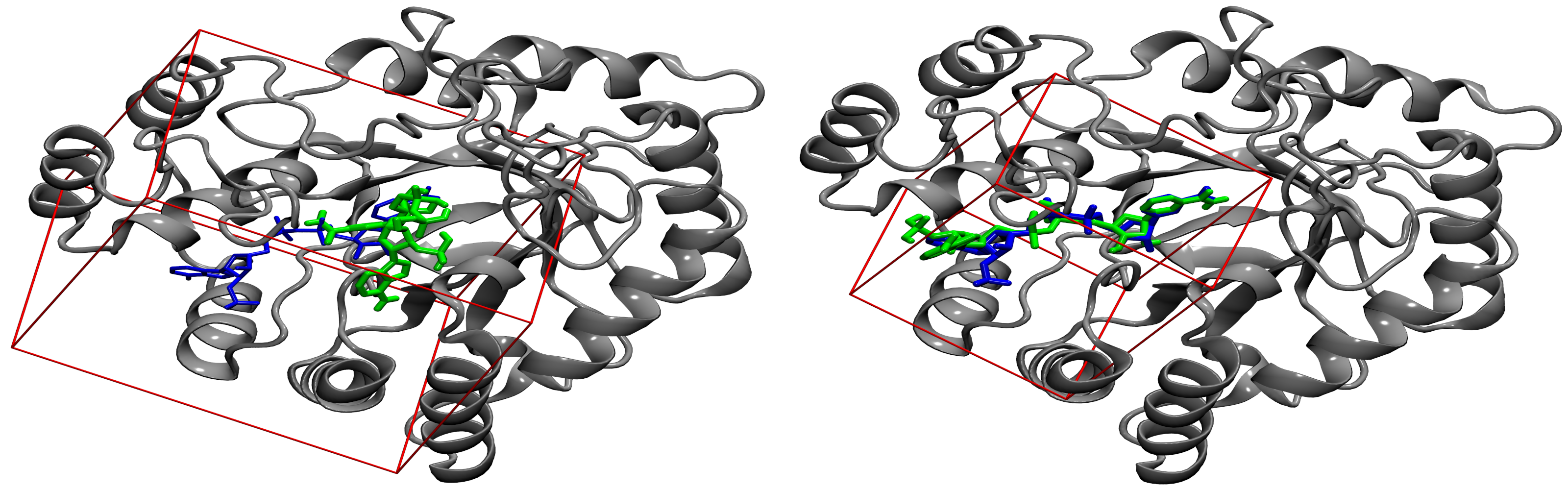
Molecular docking has profound applications in drug discovery and development. One of the critical parameters for ligand docking is the size of a search space used to identify low-energy binding poses of drug candidates. We proposed a new procedure for calculating the optimal docking box size that maximizes the accuracy of binding pose prediction and yields an improved ranking in virtual screening. Importantly, the optimized search space systematically gives better results than the default method not only for experimental pockets, but also for those predicted from protein structures. Our approach can be employed to fully automate large-scale virtual screening calculations by customizing docking protocols on the fly for individual library compounds.